Laborstudie: Forschende entwickeln Grundlage für neue Therapien frühkindlicher Epilepsien
Epileptische Enzephalopathien sind spezielle Formen der Epilepsie, die häufig durch genetische Defekte verursacht werden. Sie treten bereits bei Säuglingen auf und sind kaum behandelbar. Die Krankheitsmechanismen und die Ursachen für die schwere Behandelbarkeit dieser frühkindlichen, mitunter tödlich verlaufenden Erkrankungen sind bisher unzureichend untersucht. Forschende um Prof. Dr. Dirk Isbrandt haben nun in Studien an Mäusen gezeigt, dass bestimmte, von Patienten abgeleitete pathologische genetische Sequenzänderungen die Nervenzellen im Gehirn so verändern können, dass herkömmliche Epilepsie-Medikamente nicht wirken und die Symptome sogar verschlimmern können. Ihre Befunde sind ein wichtiger Beitrag zum Verständnis der Erkrankung und bilden die Grundlage zur Entwicklung und Testung neuer Behandlungsansätze für Therapien bei Menschen. Die Ergebnisse sind in der Fachzeitschrift eLife veröffentlicht.
Bei der Epilepsie kommt es zu Veränderungen der elektrischen Aktivität im Gehirn, die mit Krampfanfällen, Stürzen, unwillkürlichen zuckenden Bewegungen und Bewusstseinsstörungen einhergehen können. Diese Symptome hatten der Krankheit den – mittlerweile veralteten – Namen „Fallsucht“ eingebracht. Epilepsien können durch Unfälle, Infektionen, Stoffwechselerkrankungen, Gifte oder Gendefekte hervorgerufen werden. Viele Epilepsie-Erkrankungen sind heutzutage gut mit Medikamenten behandelbar, so dass die Betroffenen anfallsfrei und ohne größere Nachteile aufgrund der Erkrankung leben können – nicht aber bei speziellen Formen der Epilepsie: Den sogenannten Epileptischen Enzephalopathien, die bereits im ersten Lebensjahr und sogar ab dem ersten Tag der Geburt auftreten können: „Bei diesen frühkindlichen Erkrankungen wirken herkömmliche Anfallsmedikamente häufig nur unzureichend und manchmal sogar geradezu paradox: Sie lösen noch heftigere Symptome aus“, sagt Prof. Dr. Dirk Isbrandt, Forschungsgruppenleiter am DZNE und Direktor des Instituts für Molekulare und Verhaltensneurowissenschaften an der Universität zu Köln. „Diese bislang kaum behandelbaren Epilepsien sind oft mit einer lebenslangen Belastung durch Krampfanfälle, Entwicklungsverzögerungen, Verhaltensauffälligkeiten und Intelligenzminderung verbunden. Einige Krankheitsformen können sogar tödlich verlaufen.“
Anknüpfungspunkte für neue Therapien finden
Viele herkömmliche Medikamente gegen die häufigsten Formen der Epilepsie wirken, indem sie die „überschießende“ elektrische Aktivität von Ionenkanälen in der Hülle der Gehirnzellen dämpfen: Dadurch wird die Übererregbarkeit der Nervenzellen reduziert. Bei Epileptischen Enzephalopathien hingegen funktionieren diese Arzneimittel oft nicht zufriedenstellend. Ursache dieser speziellen Epilepsieformen sind meist Gendefekte, also Fehler im Erbgut. „Durch genetische Analysen von Epilepsiepatienten anderer Forschungsgruppen wissen wir bereits, dass sogenannte HCN-Kanäle in der Zellmembran bei bestimmten Formen dieser frühkindlichen Erkrankungen dauerhaft in ihrer Funktion gestört sein können“, so Isbrandt. „Dieser Funktionsverlust stört die Regulation der Erregbarkeit in Nervenzell-Netzwerken und hat epileptische Symptome zur Folge. Warum übliche Medikamente hier nicht helfen, sondern sogar kontraproduktiv sind, wollten wir genauer untersuchen.“ Um den Weg für neue Therapiemöglichkeiten zu bereiten, hat das Team um Isbrandt gemeinsam mit weiteren Forschenden des DZNE, der Uniklinik Köln und der Columbia Universität in New York diese Genmutationen im Modellorganismus erforscht.
Mit unserer Studie haben wir neue Modelle Epileptischer Enzephalopathien entwickelt und mit ihrer Hilfe den Krankheitsmechanismus besser verstanden.
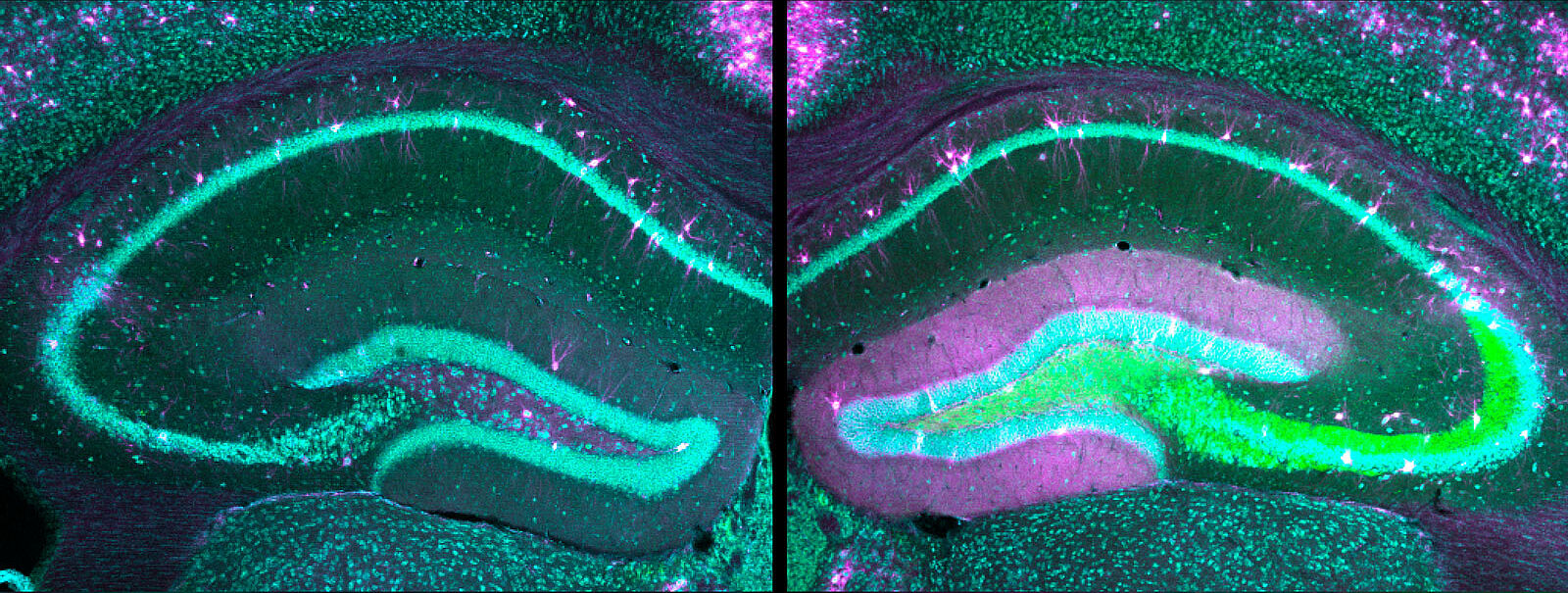
„Wir haben für unsere Studie zwei verschiedene neue Mausmodelle mit modifizierten HCN1-Genen entwickelt, von denen bekannt ist, dass sie mit Epileptischen Enzephalopathien in Verbindung stehen“, so Dr. Andrea Merseburg, Wissenschaftlerin in Dirk Isbrandts Forschungsgruppe und Erstautorin der aktuellen Studie. Mit Hilfe eines speziellen molekularbiologischen Verfahrens – der sogenannten CRISPR/Cas9-Methode – wurden in das Erbgut von Mäusen Genmutationen übertragen, die bei betroffenen Kindern vorkommen und je nach Gendefekt zu unterschiedlich schweren Formen der Erkrankung führen. „Dadurch konnten wir zwei Tiermodelle mit verschieden stark ausgeprägten Krankheitsformen erstellen“, berichtet die Neurowissenschaftlerin.
Bei der Messung der Hirnströme zeigten die beiden neu entwickelten Mausmodelle typische Störungen, die von erkrankten Menschen bekannt sind. Auch die Symptome entsprachen – abhängig vom jeweiligen Gendefekt – den milderen oder schweren Krankheitsverläufen bei betroffenen Kindern und beinhalteten neben den spontanen Anfällen auch kognitive Defizite und Verhaltensauffälligkeiten. Zudem gab es eine ähnliche Verschlimmerung der Anfälle als Reaktion auf antiepileptische Medikamente. „Wir konnten feststellen, dass die Genmutationen in den HCN-Kanälen die Nervenzellen insgesamt so stark verändern, dass herkömmliche Medikamente die Nervenzell-Netzwerke sogar noch erregbarer machen, was zu heftigen paradoxen Krampfanfällen geführt hat“, sagt Merseburg. „Mit unserer Studie haben wir neue Modelle Epileptischer Enzephalopathien entwickelt und mit ihrer Hilfe den Krankheitsmechanismus besser verstanden.“ Weitere Studien seien notwendig, damit zukünftig wirksame, gezielt auf die Krankheitsmechanismen zugeschnittene Wirkstoffe für Kinder entwickelt werden könnten.
Das Projekt wurde durch das Zentrum für Molekulare Medizin der Universität zu Köln und die Deutsche Forschungsgemeinschaft (DFG) im Rahmen des Sonderforschungsbereichs 1451 „Schlüsselmechanismen normaler und krankheitsbedingt gestörter motorischer Kontrolle” gefördert.
Originalveröffentlichung
Seizures, behavioral deficits and adverse drug responses in two new genetic mouse models of HCN1 epileptic encephalopathy.
Andrea Merseburg et al.
eLife (2022)
DOI: https://doi.org/10.7554/elife.70826