Laboratory study: Researchers develop basis for new therapies of early childhood epilepsies
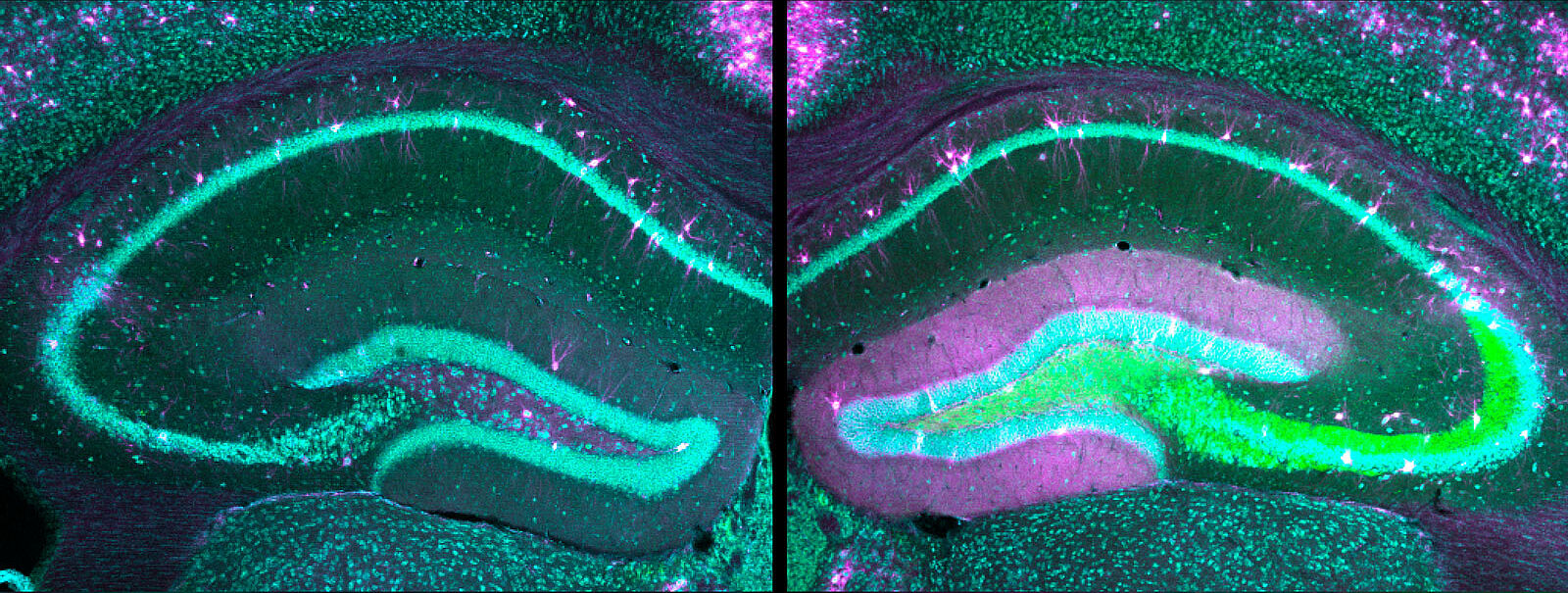
Epileptic encephalopathies are specific forms of epilepsy that are often caused by genetic defects. They manifest as early as infancy and are hardly treatable. The disease mechanisms and the question of why these early childhood disorders are challenging to treat and can also be lethal have not yet been adequately investigated. Researchers of Prof. Dr. Dirk Isbrandt’s team have now shown in studies in mice that certain patient-derived gene sequence variations can alter neuronal networks in the brain in a way that they fail to respond to conventional antiseizure medication, which can sometimes even worsen the symptoms. Their results are an important contribution to understanding the disease and form the basis for developing and testing new treatment options for human therapies. The findings are published in the journal eLife.
Epilepsy is characterized by changes in electrical activity in the brain that can be accompanied by seizures, falls, involuntary twitching movements, and impaired consciousness. Due to these symptoms, the disease was called " falling sickness" in the past, a name that has since become obsolete. Epilepsy can be caused by accidents, infections, metabolic disorders, toxins, or genetic defects. Nowadays, many epilepsy disorders can adequately be treated with medication so that patients can mostly live seizure-free and without major disadvantages due to the disease – but not in the case of particular forms of epilepsy – the so-called epileptic encephalopathies, which can occur as early as the first year of life and even from the first day of birth. "In these early childhood diseases, standard antiseizure drugs often have an inadequate effect and sometimes even a paradoxical one, that is, they trigger rather than suppress seizures," said Prof. Dr. Dirk Isbrandt, research group leader at the DZNE and director of the Institute for Molecular and Behavioral Neurosciences at the University of Cologne. "These epilepsies, which are mainly resistant to any treatment, are often associated with a lifelong burden of seizures, developmental delay, behavioral impairments, and intellectual deficits. Some forms of the disease can even be lethal."
Finding a new therapeutic approach in humans
Many conventional drugs for the most common forms of epilepsy work by dampening the excessive electrical activity of ion channels in the membrane of brain cells, reducing the hyperexcitability of neurons. In epileptic encephalopathies, however, these drugs often do not work satisfactorily. These specific forms of epilepsy are usually caused by genetic defects. "Through genetic analyses of epilepsy patients performed by other research groups, we know that the so-called HCN channels in the cell membrane can be permanently impaired in their function in these early childhood diseases," said Isbrandt. "This loss of function disrupts the regulation of excitability in neuronal networks and results in epileptic symptoms. Why common medications do not help here but are even counterproductive is what we sought to investigate in more detail." To pave the way for new therapeutic options, Isbrandt's team joined forces with other researchers from the DZNE, the University Hospital of Cologne, and Columbia University in New York to research these gene mutations in the model organism.
With our study, we have developed new models of epileptic encephalopathies and used them to gain a better understanding of the disease mechanism.
"For our study, we generated two different mouse models with modified HCN1 genes that are known to be associated with epileptic encephalopathies," said Dr. Andrea Merseburg, a scientist in Dirk Isbrandt's research group and lead author of the current study. Using a specific molecular biology method – the so-called CRISPR/Cas9 gene editing technology – pathological sequence variants that occur in affected children and lead to disease forms of varying severity, depending on the gene defect, were transferred into the genome of mice." This allowed us to create two animal models with different forms of disease severity," the neuroscientist explained.
When electrical brain activity was measured, the two newly generated mouse models showed typical changes known from affected humans. The symptoms also matched the disease's milder or more severe progressions in affected children – depending on the genetic defect – and included cognitive deficits, behavioral impairments, and spontaneous seizures. In addition, there was a similar exacerbation of seizures in response to antiepileptic medications. "We found that the gene mutations in the HCN channels altered the neurons to such an extent that standard drugs increased the excitability of the neuronal networks, leading to severe paradoxical seizures," Merseburg said. "With our study, we have developed new models of epileptic encephalopathies and used them to gain a better understanding of the disease mechanism." Further studies are needed, she added, so that effective drugs targeting the identified disease mechanisms can be developed for children in the future.”
The project was funded by the Center for Molecular Medicine Cologne (CMMC) at the University of Cologne and the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) as part of Collaborative Research Centre 1451 “Key Mechanisms of Motor Control in Health and Disease.”
Original publication
Seizures, behavioral deficits and adverse drug responses in two new genetic mouse models of HCN1 epileptic encephalopathy.
Andrea Merseburg et al.
eLife (2022)
DOI: https://doi.org/10.7554/elife.70826