
Forschungsschwerpunkte

Die frontotemporalen Lobärdegenerationen (FTLD) und amyotrophe Lateralsklerose (ALS) weisen wie die meisten anderen neurodegenerativen Erkrankungen charakteristische Merkmale auf, nämlich das Auftreten abnormaler Proteinaggregate, die charakteristische zelluläre Einschlüsse im Gehirn bilden, und das Vorhandensein von vererbten Formen der Krankheit, die durch Mutationen in spezifischen Genen verursacht werden.
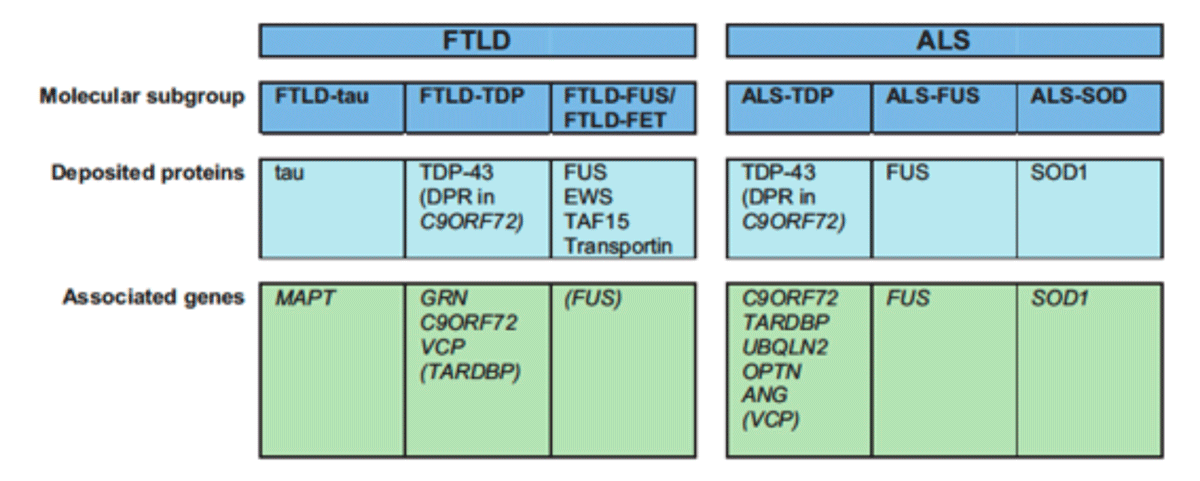
Unser Verständnis der molekularen Grundlagen der FTLD und ALS hat sich im letzten Jahrzehnt dramatisch verändert mit den bahnbrechenden Entdeckungen von TDP-43 und FUS als neue aggregierende Krankheits-Proteine bei ALS und FTLD (Abbildung 1) und der Identifizierung neuer Krankheitsgene (zB Progranulin und C9orf72 ). Dies ermöglicht es nun, fast alle FTLD und ALS-Fälle in drei unterschiedliche molekulare Gruppen zu klassifizieren (Abbildung 2). Die Identifizierung von gemeinsamen Krankheitsproteinen und -genen bei FTLD und ALS liefert einen eindeutigen Beweis auf molekularer Ebene, dass Subgruppen von FTLD und ALS gemeinsame Pathomechanismen aufweisen. Darüber hinaus impliziert die physiologische Rolle der aggregierenden Proteine TDP-43 und FUS als DNA / RNA bindende Proteine einen RNA-Fehlmetabolismus als Schlüsselereignis in der Krankheitsentstehung.
Ziele unserer Forschung sind nun die physiologischen Funktionen von TDP-43-, FUS- und C9orf72-Proteinen im zentralen Nervensystem zu entschlüsseln, die Mechanismen zu klären, die dem TDP-43 und FUS-bedingten Zelltod zugrunde liegen (loss of functions vs. Toxic gain of functions) und molekulare Ähnlichkeiten und Unterschiede von TDP-43 und FUS zwischen ALS und FTLD zu analysieren. Dieses Wissen wird entscheidend für die Entwicklung zukünftiger therapeutischer Ansätze und Biomarker sein.
Um dies zu erreichen, fokussieren wir auf die Analyse von postmortalen menschlichen Gewebeproben, die in unserer Hirngewebebank (Brain-Bank) gesammelt werden und auf die Erzeugung und Charakterisierung von neuen genetisch modifizierten Mausmodellen unter Verwendung von Knock-Out-, Knock-In- und Überexpressionsansätzen.
Wir setzen ein breites Methodenspektrum ein mit modernsten histologischen und proteomischen Techniken, Bildanalyseverfahren, Antikörpergenerierung, Zellkultur (primäre Zellkulturen, organotypische Schnittkulturen) sowie molekularbiologischen Techniken.
Derzeitige spezifische Projekte in unserem Labor:
- Identifizierung posttranslationaler Modifikationen von TDP-43- und FET-Proteinen in FTLD / ALS-Subtypen und deren pathomechanische/physiologische Relevanz
- Entwicklung neuartiger genetisch modifizierter Mausmodelle und Modellsysteme (z. B. organotypische Schnittkulturen), um wesentliche pathologische Aspekte von menschlichen TDP-43- und FUS Krankheiten nachzuahmen.
- Identifizierung von transkriptomischen / epigenetischen / proteomischen Veränderungen im murinen ZNS nach konditioniertem TDP-43 und FUS-Knock-out und Validierung ihrer funktionellen und pathogenetischen Relevanz.
- Charakterisierung der physiologischen Funktionen von C9orf72 im ZNS und dessen Zusammenspiel mit TDP-43.