
Areas of investigation/research focus

Frontotemporal lobar degenerations (FTLD) and amyotrophic lateral sclerosis (ALS) share characteristic features with most other neurodegenerative diseases, namely the presence of abnormal protein aggregates forming characteristic cellular inclusions in the brain and the presence of inherited forms of the disease caused by mutations in specific genes.
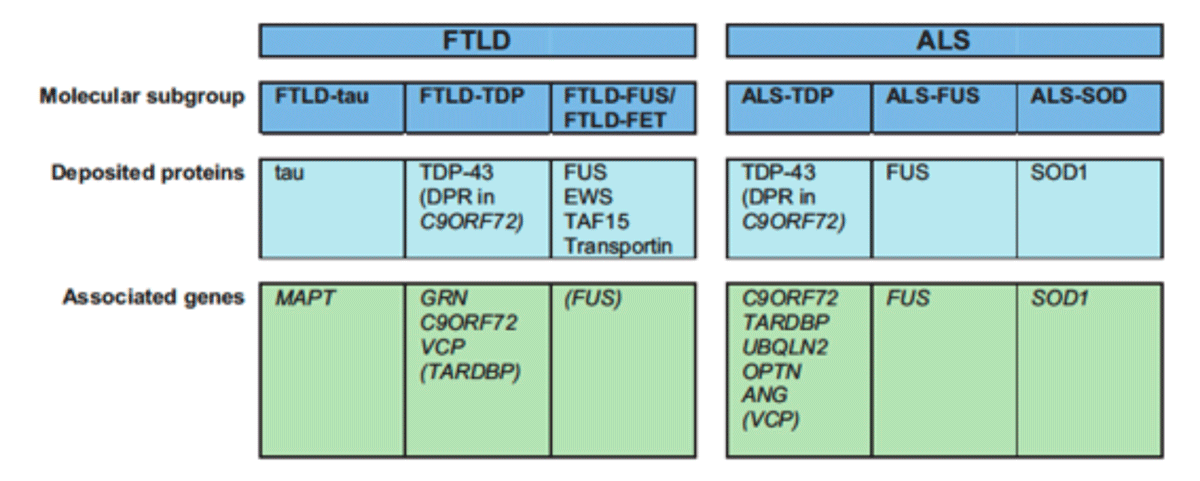
Our understanding of the molecular basis of FTLD and ALS has changed dramatically within the last decade with the seminal discoveries of TDP-43 and FUS as novel ALS/FTLD disease proteins (Figure 1) and the identification of new disease genes (e.g. progranulin and C9orf72). This allows now to classify almost all FTLD and ALS cases into 3 distinct molecular groups (Figure 2). The identification of common disease proteins and genes in FTLD and ALS provided clear molecular evidence that subsets of FTLD and ALS share common pathomechanisms. Moreover, the physiological role as DNA/RNA binding proteins of the aggregating proteins TDP-43 and FUS implies RNA mis-metabolism as key event in disease pathogenesis.
Our main research goals are now to decipher the physiological functions of TDP-43, FUS and C9orf72 proteins in the central nervous system, to elucidate the mechanism(s) underlying TDP-43 and FUS-related cell death (loss of functions vs. Toxic gain of functions) and to dissect molecular similarities and differences of TDP-43 and FUS between ALS and FTLD cases. This knowledge will be crucial for the development of future therapeutic approaches and biomarkers.
To achieve this, we focus on the analysis of postmortem human tissues collected in our brain bank and the generation and characterization of novel genetically-modified mouse models using knock-out, knock-in and overexpression approaches.
We apply a broad methodological spectrum including cutting edge histological and proteomics techniques, image analysis, antibody generation, primary neuron and organotypic slice cultures and molecular biology approaches.
Current specific projects in our lab include:
- Identification of posttranslational modifications of TDP-43 and FET proteins in FTLD/ALS subtypes and their pathomechanistic/physiological relevance
- Development of novel genetically modified mouse models and models systems (e.g. organotypic slice cultures) to mimic essential pathological aspects of human TDP-43 and FUS diseases.
- Identification of transcriptomic/epigenetic/proteomic changes in the murine CNS upon conditional TDP-43 and FUS knock-out and validation of their functional and pathogenetic relevance.
- Characterization of the physiological functions of C9orf72 in the CNS and its interplay with TDP-43.