
Research areas
The research focus of my group is to study pathomechanisms underlying neurodegenerative diseases with particular focus on amyotrophic lateral sclerosis (ALS) and Parkinson's disease. We aim to understand why neuropathology, starting from a local focus, spreads to different areas. In vitro, in vivo, and with human patient material, we investigate protein aggregation mechanisms, inflammatory processes, and gene expression regulation processes at the single cell level. We hope to understand why individual cell populations are particularly vulnerable and which factors play a role in this. In particular, we focus on the aggregating proteins alpha synuclein and TDP43.
For alpha synuclein we could already show in in vivo models that a transfer of alpha synuclein oligomers from neurons to neurons or even to non-neuronal populations takes place. Currently, we are also investigating the spread of TDP43 pathology in an ALS in vivo model. Besides investigating spreading mechanisms in PD and ALS, we are also interested in epigenetic changes in ALS or PD patient blood or human brain tissue analyzed by multiomic single cell sequencing (mRNA and ATAC sequencing).
Our new insights could have direct impact on clinical diagnostics and provide new therapeutic intervention options in Parkinson's disease, amyotrophic lateral sclerosis and other neurodegenerative diseases.
Protein Aggregation and Prion-like Propagation
Toxic protein aggregation plays a significant role in the development and progression of various neurodegenerative diseases. In these conditions, specific proteins misfold and aggregate, leading to the formation of toxic protein aggregates that disrupt cellular functions and contribute to neuronal damage and cell death.
Besides intracellular protein aggregation, prion-like spreading of disease related proteins or aggregates has been observed in several neurodegenerative diseases. Protein aggregates, e.g. alpha synuclein (a-syn), propagate and spread between cells, contributing to the progression of the disease.
Aging related mechanisms: Interaction between protein aggregation, age and exposure time to misfolded proteins
Aging is a significant risk factor for the development of neurodegenerative diseases. As individuals age, their cells and tissues undergo various changes, including a decline in cellular repair mechanisms, increased vulnerability to oxidative stress, and changes in protein homeostasis. These age-related processes can contribute to the accumulation or aggregation of proteins and cellular dysfunction, making the brain more susceptible to neurodegenerative conditions. Within the CRC 1506 “Aging at Interfaces” we want to understand whether cellular aging with all its associated altered mechanisms is the prerequisite for protein aggregation in general or whether the exposure time to misfolded proteins determines the accumulation of protein aggregates with their detrimental consequences. Further we want to understand whether pathological age-related mechanisms can be rescued by manipulation of structural cellular elements.
Parkinson’s disease (PD) is the second most common neurodegenerative disease with age being the strongest risk factor. Aggregation and oligomerization of a-syn play a central role in the development of PD. Recently, we identified an age dependent accumulation of a-syn oligomers accompanied by neuronal cell loss and diminished motoric abilities using a PD mouse model based on human a-syn protein complementation expressing α-synuclein split Gaussia luciferase (called S1/S2 model). This model allows highly sensitive assessment of a-syn oligomers since it is based on a human a-syn protein complementation assay that expresses a-syn split Gaussia luciferase. This mouse model is characterized by accumulation of a-syn oligomers accompanied by neuronal cell loss and diminished motoric abilities, as well as trans-cellular spreading of a-syn oligomers.
(Please see Image 1 below and click on the image to enlarge it.)
The underlying mechanisms responsible for an accumulation of a-syn oligomers during aging are not clear to date. The aim of this project is to understand whether neurons at a certain age are most vulnerable for a-syn oligomerization or whether the exposure time to those oligomers is responsible for the detrimental effects on neurons. We therefore induce a-syn oligomer expression for four months at different ages in our S1/S2 model and compare them with mice expressing a-syn oligomers from birth regarding their motoric abilities. To determine whether motoric alterations correlate with a-syn oligomer load, we characterize a-syn oligomer species with biochemical methods. To get further insights into the underlying mechanism we are studying protein degradation mechanisms (autophagy and proteasomal activity), neurodegeneration, DNA damage, translational accuracy as well as perform single cell transcriptomics. Since we observed previously that a-syn oligomers can spread from neurons expressing a-syn to other neurons not expressing a-syn we want to determine whether cell to cell spreading is an age-related mechanism. To study cell-to-cell transmission on a single cell resolution we employ visium spatial transcriptomics and further investigate the consequences of a-syn spreading.
(Please note Image 2 and click on the image to enlarge it.)
During aging, the activity of protein Cell division cycle 42 (Cdc42) increases in blood, liver and brain cells of the mouse. It has been shown that the increase of Cdc42 activity leads to disorganization in cells like hematopoietic stem cells and with this contributes to changes in age. Interestingly, a-syn oligomers can bind specifically to Cdc42 effector proteins. Recently, it has been shown that inhibition of Cdc42-activity by a specific inhibitor results ex and in vivo in a reorganization and rejuvenation of hematopoietic stem cells. Since Cdc42 activity is increasing with age, the question is whether modulation of Cdc42 activity might rescue the age-dependent Parkinson’s disease phenotype via changes on a-syn oligomerization. We are therefore pharmacologically target Cdc42 in PD mice at different ages to test whether the PD phenotype and a-syn oligomers can be ameliorated by Cdc42 modulation.


Trauma: Effects of peripheral and CNS trauma on the brains of healthy and Parkinson´s Disease mouse models
Despite intensive research over the past decades, the interaction of trauma and its consequences on brain function and initiation of neurodegenerative diseases is not sufficiently unraveled. A link between traumatic injuries and their effects in the brain has been suggested, however, the exact cellular processes involved, as well as the mechanisms of how this happens are still mostly unknown. As trauma has been implicated in the development of Parkinson’s disease (PD) we aim to unravel within the CRC 1149 “Danger Response, Disturbance Factors and Regenerative Potential after Acute Trauma” the underlying mechanisms linking trauma and inflammation to neurodegeneration.
On the molecular level, brain injury has been suggested to facilitate a-synuclein (a-syn) aggregation, which is the key protein in PD and the main pathological hallmark in the PD brain. Increased levels of a-syn were reported in the cerebrospinal fluid of patients after TBI and a-syn immunoreactive neurons were seen in resected cortical tissue after a severe TBI in humans. Besides TBI also traumatic injuries and inflammation in the periphery increase the risk of developing neuropsychiatric and neurodegenerative diseases. However, our knowledge on the pathophysiology linking peripheral inflammation to brain malfunction is still limited. We could show that a peripheral injury like a thoracic trauma leads to a strong increase in a-syn oligomers while the total a-syn amount remained constant, suggesting that peripheral injury influences protein aggregation in the brain of PD mice.
Together with the group of Leda Dimou we are aiming to understand to what extent a TBI or a sepsis would change the physiological function of the brain regarding gliosis, adult neurogenesis, protein aggregation, a-syn spreading and neurodegeneration. Additionally, we want to answer whether a preceding trauma can exacerbate or even trigger PD pathology on the long term, not only on a cellular level but also regarding behavioral changes of affected mice. On the other hand, we are asking how a comorbidity such as PD can alter the outcome of a traumatic lesion at short timepoints after an injury.

Molecular chaperones: Heat shock proteins and TDP43
In 10 % of ALS cases, the disease is inherited and caused by mutations in different genes such as C9ORF72, SOD-1 and TARDBP. TARDBP encodes the transactive response DNA-binding protein 43 kDa (TDP-43), which seems to play a critical role in ALS. 97 % of all ALS cases, independent of the familial background, show TDP-43 positive cytoplasmic inclusions. In ALS, TDP-43, normally predominantly located in the nucleus, mislocalizes in the cytoplasm and forms cytoplasmic TDP-43. Therefore, protein misfolding and aggregation seems to be a critical process in the cause of the disease. Heat shock protein 70 kDa (HSP70), a molecular chaperon, has been identified to be neuroprotective in several neurodegenerative diseases. For ALS, it has been reported that HSP70 regulates TDP-43 aggregation in the heat shock response and could prevent aggregation of the 25-kDa C-terminal fragment of TDP-43. The aim of this project is to study the effect of HSP70 family members on TDP-43 aggregation, localization and solubility. We focus not only on Hsp70, but also on neuron specific Hsp proteins. To this end we employ TDP-43 cell culture models as well as patient cells and study TDP43 aggregation, localization as well as binding and interaction of TDP-43 and HSP70 family members.

Development and characterization of novel TDP-43 based ALS mouse models
Since TDP-43 aggregation is the main neuropathological hallmark of ALS we aim to develop mouse models that allow a quantitative and spatial resolution of TDP-43 aggregation.
To this end we have generated transgenic mouse lines containing human WT TDP-43 tagged with hGaussia luciferase or split venus YFP, called TDP-43-Luci or TDP-43-V1/V2 (TV1/TV2), respectively. TDP-43 is fused to either N- or C-terminal non fluorescent Venus YFP that reconstitute to fluorescent Venus VFP upon aggregation. To aim towards inducible and cell type specific TDP43 expression, we employ a Tamoxifen inducible Cre-ERT2 system.
With these models we will study the potential correlation between TDP-43 aggregation and an ALS phenotype, as well as in vivo TDP-43 aggregation and propagation in a quantitative as well as spatial resolution. We pay particular attention to TDP-43 transmission from neuronal to neuronal cells as well as also to non-neuronal cells and study its subsequent consequences.

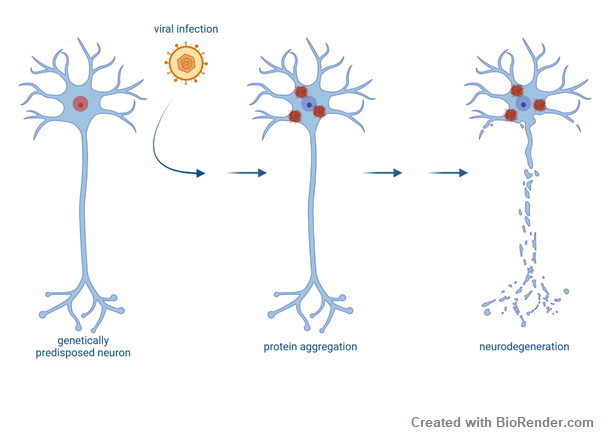
Viral infections and ALS
Development of ALS is likely caused in a multi-step process. External factors might act on top of a genetic predisposition to trigger disease onset. In this project we study whether viral infections and the subsequent antiviral immune response impact ALS pathogenesis. Viral infections might have either happened in early adulthood or later in life. Whether this is clinically and epidemiologically linked to ALS needs to be explored. On a molecular level, viral infections have recently been shown to induce stress granules and FUS proteinopathy in ALS models. Therefore, we also aim elucidate together with the group of Konstantin Sparrer the molecular mechanisms underlying viral infections on TDP43 aggregation as potential risk factors for development and/or progression of ALS.
People







(Neuro)Inflammation
Immune system mechanisms in PD and ALS
The immune system offers several unique chances for the research of neurodegenerative diseases: biomarker research, investigation of disease mechanisms, potential therapeutic targets, and translational insights. In this project, we study the interface between the immune system and neurodegenerative diseases by a combination of modern bioinformatic and machine-learning techniques with classical molecular biology techniques from the fields of immunology and neuroscience. As an easily accessible, highly-reactive ‘mirror’ of systemic processes, the immune system reflects changes in the CNS that can be measured in peripheral blood. We characterize the composition of blood cells, their gene expression, epigenetic changes and functional aberrations in PD and ALS with flow cytometry, primary cell culture and multi-omic approaches in bulk and single-cell genomics (ATAC-seq, RNA-seq, protein expression). To search for novel disease mechanisms and potential therapeutic targets, we characterize the circulating T cells and their T-cell receptor with flow cytometry, ELISPOT, T-cell receptor sequencing and machine learning; we test the effect of recombinant antibodies to CNS antigens on protein aggregation with in-vitro assays; finally, we investigate the receptors and cytokines involved in innate immunity to aggregated proteins with ELISA and molecular biology techniques.

The interaction of Parkinson’s disease, stress and inflammation
The hypothalamic-pituitary-adrenal axis (HPA) axis is one of the two major stress systems, that is activated in response to stress. Recent studies suggest that α-syn aggregation may impact the functioning of the HPA axis. Reduced ACTH levels have been observed in PD patients and altered levels of cortisol in PD patients have been reported.
In this project we aim to study the impact of α-syn on the stress-associated axis of the immune system and the glucocorticoid (GC) sensitivity of immune cells. This project is performed in collaboration with the group of Stefan Reber, who has a strong expertise in stress related changes in the immune system. Towards this end we characterize stress parameters in our PD a-syn mouse model as well as in human peripheral blood mononuclear cells (PBMCs) of PD patients.

Autoantibodies in PD and ALS
Autoantibodies are antibodies produced by the immune system that mistakenly target and attack the body's own tissues or proteins. Recently, autoantibodies have been identified in some cases of patients with neurodegenerative diseases, which potentially contribute to the progression or exacerbation of neurodegenerative disorders. However, the role of autoantibodies in neurodegenerative diseases and the understanding of their involvement is not clear to date.
We aim to look for circulating auto-antibodies to known and putative CNS antigens and investigate the complement activation. It is already known that the proteins TDP-43 and SOD1 are directly related to ALS disease. Especially the pathologically modified forms in the sense of aggregation and changes in localization play a role.
Therefore, in this project we investigate detection, quantitative determination and comparative presentation of autoantibodies against ALS disease associated proteins in the human serum of ALS patients and healthy control subjects. We also want to study whether extracellular aggregate formation is possible and to what extent this process can be inhibited by the addition of antibodies.
Immunophenotyping in Major Depressive Disorder (MDD) and schizophrenia
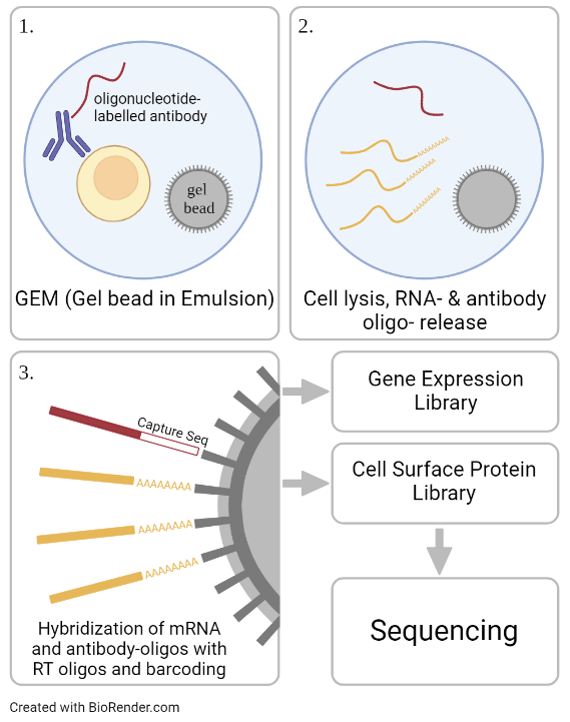
In a subset of patients, MDD is associated with inflammatory processes. To further investigate this relation, we aim to immuno-characterize peripheral blood mononuclear cells (PBMCs) from MDD patients and healthy controls using cellular indexing of transcriptomes and epitopes (CITE-Seq). Thereby, differentially expressed genes as well as changes in the cell surface proteome can be investigated on a single cell level. Additionally, extracellular vesicles are extracted from the patient’s plasma and their cargo is analyzed using mass spectrometry for further immune phenotyping as well as biomarker discovery. Structural and activity related changes have been detected in distinct brain regions of MDD patients, including the amygdala and the anterior cingulate cortex, areas that are important for cognitive and affective functions. In a multi-omic approach, chromatin accessibility and transcriptomics are analyzed on a single cell level in post mortem brain samples of MDD patients.
People

(Epi) Genetics
Epigenetic Regulation of Gene Transcription in Amyotrophic Lateral sclerosis, FTD and psychiatric diseases
During life, our organism is exposed to many different risk factors which induce epigenetic changes that finally contribute to the regulation of gene expression. An insight into the cumulative effect of DNA and histone modifications provide the chromatin landscape. Furthermore, chromatin plays a critical role in the regulation of gene expression, as only accessible genomic regions allow the transcriptional machinery to interact with the DNA.
For several neurodegenerative diseases epigenetic mechanisms have been proposed to be involved.
To investigate a mechanistic link between chromatin accessibility and ALS disease pathogenesis, we use a multi-omic approach combining an assay for transposase accessible chromatin using sequencing (ATAC-seq) and RNA-seq at bulk and single-cell level in peripheral blood mononuclear cells (PBMCs) from sporadic ALS patients and healthy controls, and integrate these results by thorough data science analysis.
We were able to show that a genome-wide signature of ALS (‘epiChromALS’) can be found in the chromatin accessibility of PBMCs from sporadic ALS patients. Functional enrichment analysis of these differentially accessible peaks revealed an association with neuronal functions and neuronal differentiation in the less accessible genomic regions, suggesting that neurodevelopmental processes are epigenetically impaired in ALS. These common epigenetic marks were associated with neuronal terms, suggesting that they are systemic.
To further corroborate our findings of a peripherally detectable epigenetic chromatin accessibility signature, we set up a larger scaled study where we investigate the chromatin accessibility in combination with gene expression profiling in single-nuclei of post-mortem tissue from the areas most affected in ALS, such as the motor cortex and spinal cord. Dysregulated epigenetic patterns in different cell types and tissues and their effects on gene expression programs are still underexplored, although they provide new insights into cell-specific and systemic alterations. Especially the understanding of systemic changes may open new doors for the development of new therapies focusing on the design of epigenetic drugs.


People

Biomarker
Contactin-associated protein-like 4
MDD is a complex and highly heterogeneous disease and the underlying pathomechanisms are most likely multifactorial and not fully understood to date. Contactin-associated protein-like 4 (CNTNAP4) belongs to a subgroup of the neurexin superfamily and is expressed in the CNS in excitatory as well as inhibitory neurons and oligodendrocytes. In neurons, CNTNAP4 is localized presynaptically and contributes to cell adhesion and dopaminergic as well as GABA-ergic signal transmission. Previously, a proteomic analysis of cerebrospinal fluid of psychiatric patients detected changes in protein levels of MDD patients compared to healthy controls. Significantly reduced concentrations were found for eight proteins, one of them being CNTNAP4, making it a potential biomarker for MDD. We therefor aim to validate the biomarker applicability of the protein. Furthermore, CNTNAP4 deficiency has been reported to deteriorate PD phenotype in asyn models. We therefore also study the mode of action of CNTNAP4 deficiency on asyn aggregation in neuronal cultures and potential pathological consequences in neurons.

Neurofilament light chain
Elevated levels of Serum Neurofilament light chain (NF-L) have already been detected in blood and CSF of pre-symptomatic and symptomatic ALS patients. This project aims at demonstrating this elevation in ALS mouse models in conjunction with other cellular changes typical for the disease. We could previously demonstrate an age- and genotype-specific increase in TDP-43 transgenic mice with reduced numbers of motoneurons as well as denervation of the neuromuscular endplates (NMJs). We want to understand why NF-L is released in the extracellular space and model this in neuronal cultures.
People

Bioinformatics
In our group we apply a wide range of multi-omics techniques, machine learning approaches and advanced statistical modelling to analyze multimodal sequencing data. We employ droplet-based single-cell sequencing to gain insights into cellular heterogeneity in tissues and identify novel cellular states in disease. We further use this technique to define new marker genes associated with a cell type and/or disease phenotype and define subpopulations within known cell types based on the gene expression profile. We also utilize bulk RNA and ATAC sequencing as well as ChIP-seq. To gain spatial information on a single-cell level we use the Visium Spatial platform from 10X Genomics and combine immunohistochemistry with transcriptomic data.
Using these techniques, we aim to uncover underlying disease mechanisms with primary patient material ranging from PBMCs to spinal cord and brain tissue and in α-syn and TDP-43 mouse models.
The utilization of multi-omics sequencing analysis, particularly at a single-cell resolution, offers a more precise understanding of the disease-related changes at the molecular level. The goal is to identify key molecular signatures, pathways, and cell types affected by e.g. the overexpression of α-syn at different ages or pharmacological treatments. Furthermore, we hope to elucidate the mechanisms behind PD and ALS to increase the number of biomarkers and therapeutics for patients in our future projects using multi-omics sequencing approaches.

People