
Areas of investigation/research focus
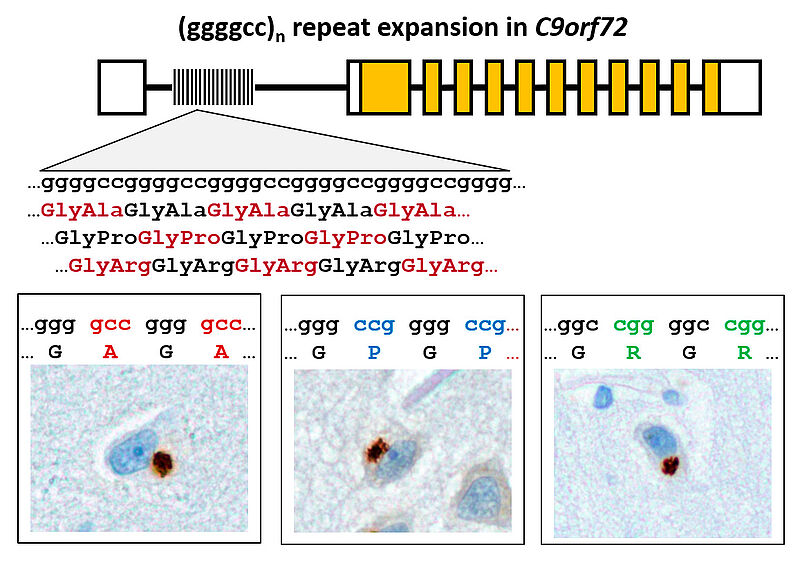
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are fatal neurodegenerative diseases with overlapping genetics and pathology. The most common cause is expansion of a (GGGGCC)n repeat in the first intron of the gene C9orf72. We discovered that the repeat region is translated in all reading frames into aggregating dipeptide-repeat (DPR) proteins (poly-GA, -GP, -GR, -PA and -PR) despite its intronic localization and lack of an ATG start codon (Mori&Weng et al., Science 2013). Funded by the ERC, the NOMIS foundation and the SyNergy excellence cluster (DFG), we are now analyzing the role of DPR proteins in pathogenesis from the ultrastructural to patient level in order to inhibit DPR synthesis and/or toxicity and thus prevent or treat C9orf72 ALS/FTD. Our cellular models show that DPR aggregates sequester key cellular proteins leading to toxicity (May et al., Acta Neuropathol 2014). The hydrophobic DPR proteins poly-GA, -GP and -PA are transmitted between cells (Zhou et al., EMBO Mol Med 2017). Since antibodies inhibit poly-GA uptake and aggregation in vitro, we are now testing immunotherapy in our mouse models. Using immunoassays, we detected poly-GP in the CSF of C9orf72 ALS and FTD patients (Lehmer et al., EMBO Mol Med 2017). Surprisingly, poly-GP levels are similar in presymptomatic and symptomatic C9orf72 carriers, indicating that DPR proteins play an early role in pathogenesis and may trigger TDP-43 pathology and neurodegeneration in a cascade-like mechanism.
We are dissecting C9orf72 pathogenesis further using primary neuron culture, mouse models (with B. Wefers, DZNE Munich), proteomics (with F. Meißner and M. Mann, MPI Martinsried) and cryo-electron tomography (with R. Fernandez-Busnadiego and W. Baumeister, MPI Martinsried). Whenever possible, we use human brain tissue to confirm our in vitro in patients (with T. Arzberger, DZNE Munich). Using cellular assays, we aim to understand the mechanisms of the unusual translation of the expanded C9orf72 repeat to develop inhibitors for future therapy.
The current data on C9orf72 pathogenesis highlights the role of RNA-metabolism and protein degradation. Importantly, these pathways are also affected by pathogenic mutations in several other known ALS/FTD-causing genes, but it is unclear how any of these mutations trigger the stereotypic TDP-43 pathology found in ~90% of all ALS cases. Therefore, we are also studying other confirmed and suspected ALS/FTD-causing mutations hoping to identify shared molecular mechanisms that lead to TDP-43 pathology, and ultimately neurodegeneration and disease. For example, we recently found that TDP-43 regulates the trafficking of recycling endosomes and subsequently affects neurotrophic signaling (Schwenk et al., EMBO J 2016).